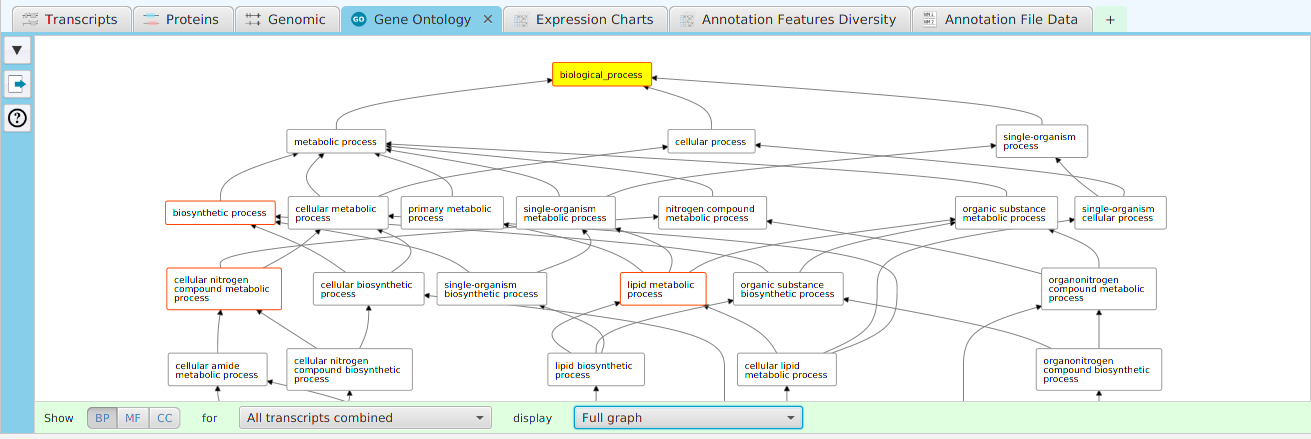
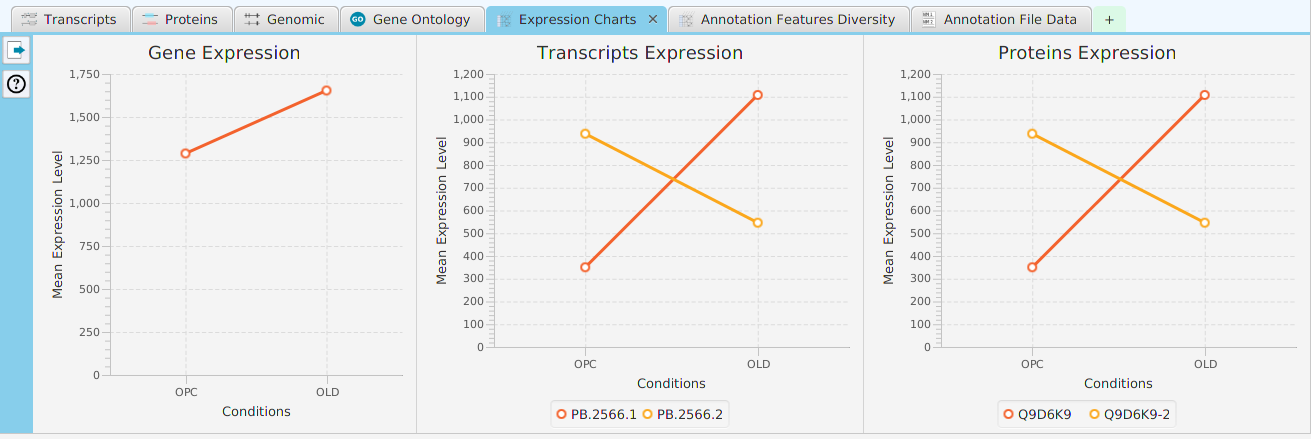
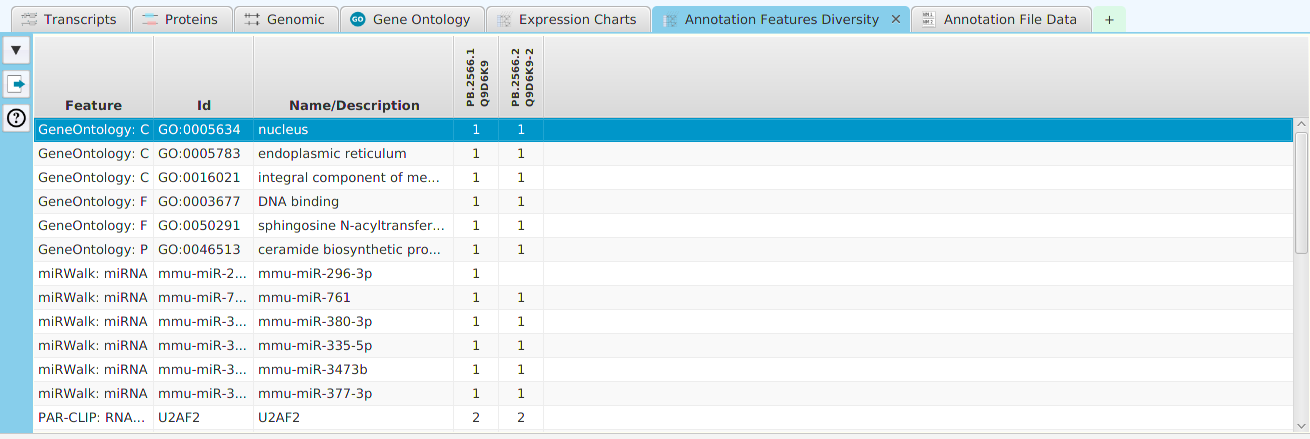
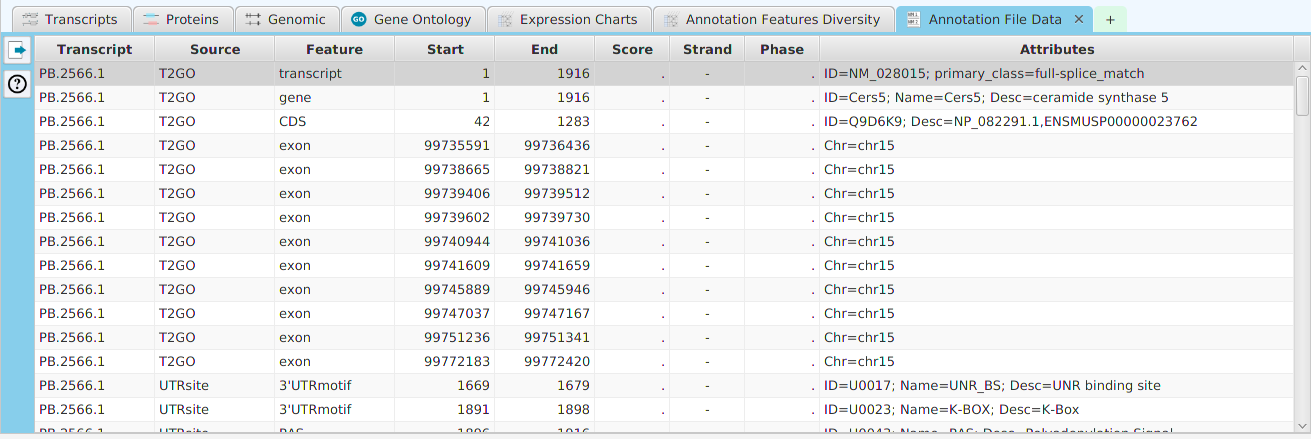
Enrichment Analysis
Enrichment analysis…
Note: Will include content from the research paper, once it becomes available
Functional Enrichment Analysis (FEA)
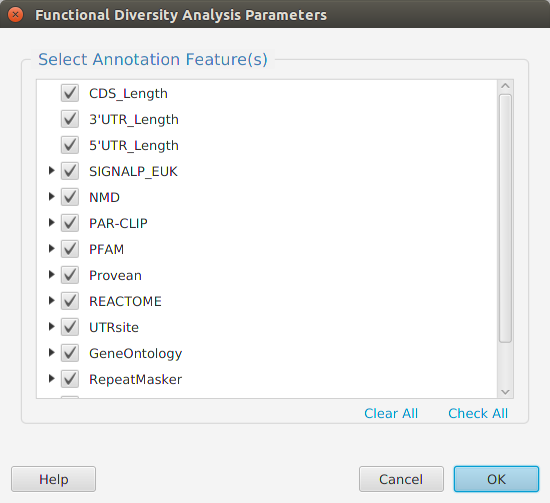
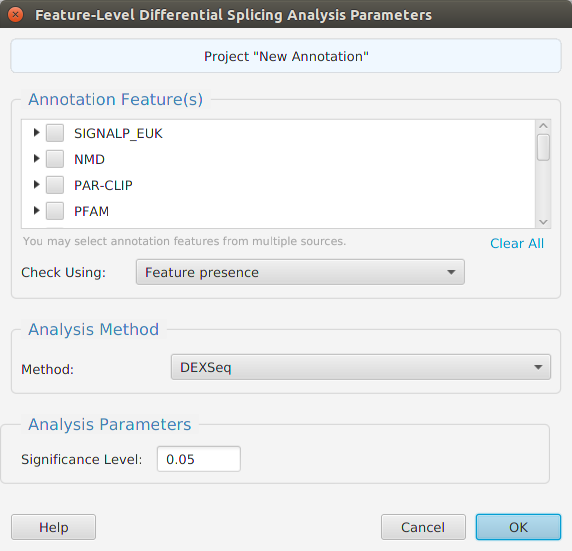
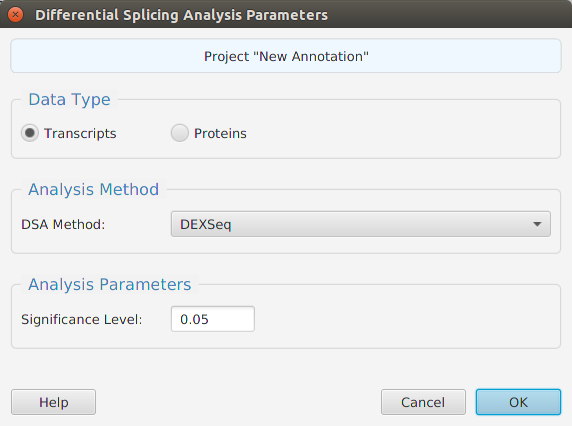
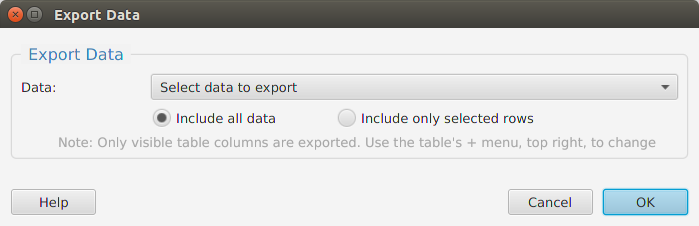
Functional enrichment analysis… You may specify what expression data type to use – genes, proteins, or transcripts – for the analysis, and the corresponding test and background lists to use. Available application lists are provided but you may use any previously generated list file. You also need to specify what annotation features to test for and some analysis package required parameters.
Note: Will include content from the research paper, once it becomes available
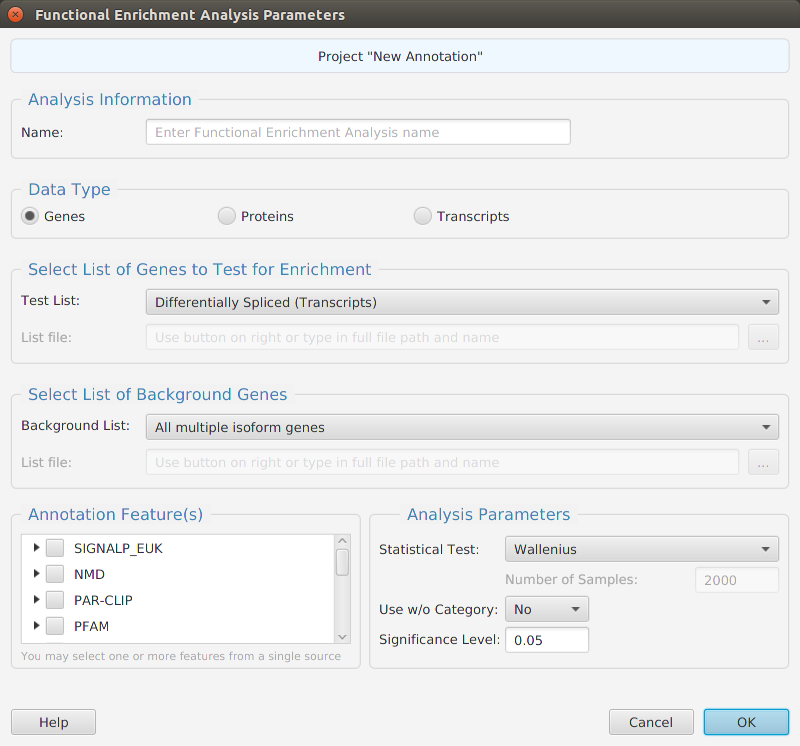
When using the application, all FEA parameters are described in the Help page which can be accessed via the Help button located on the bottom left of the dialog window.
goseq
Goseq provides “…”.You may view the documentation and installation instructions at:
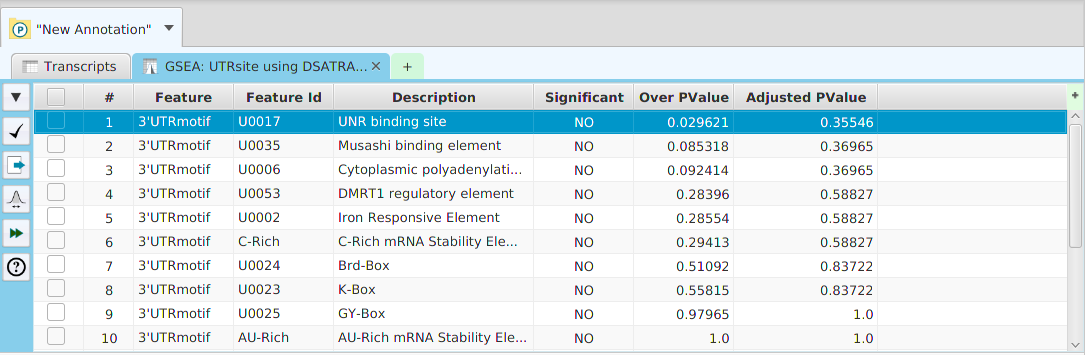
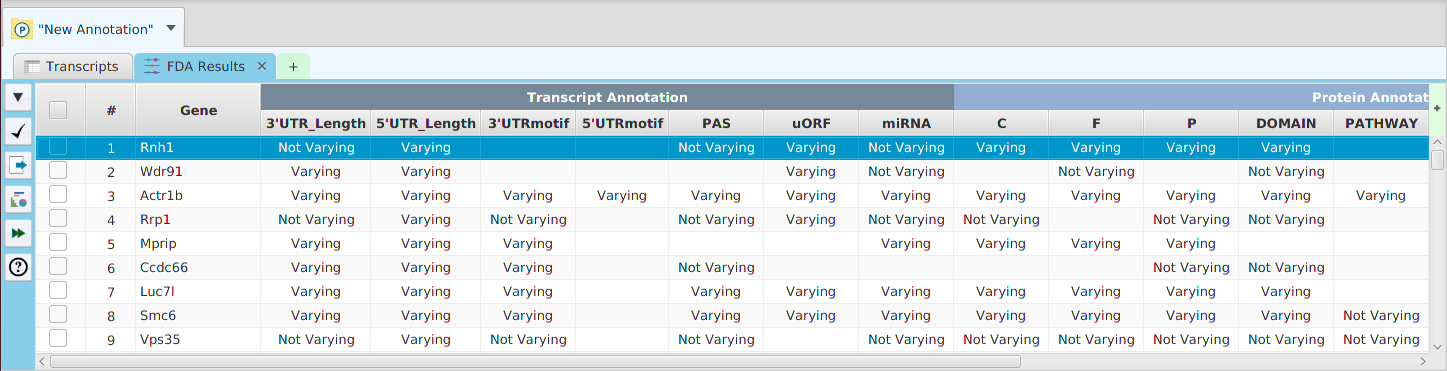
FEA Results
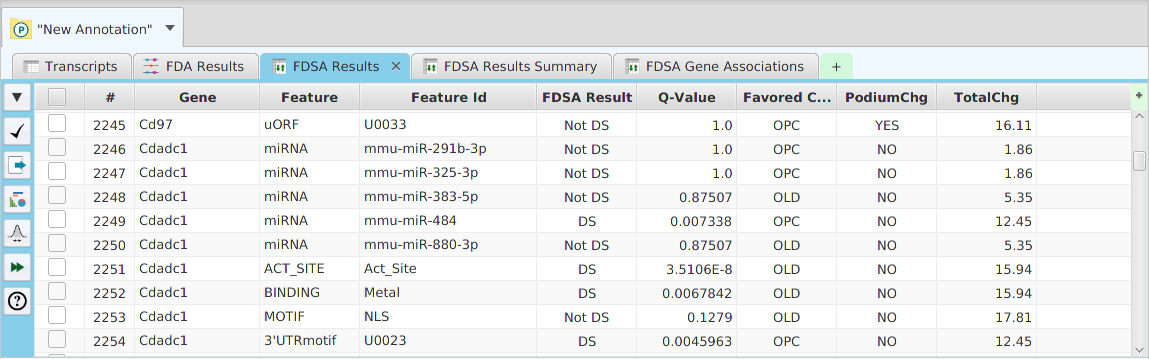
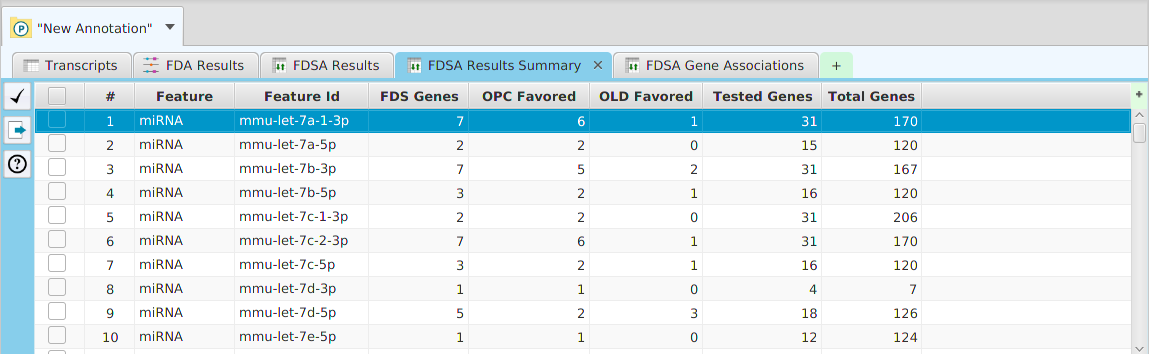
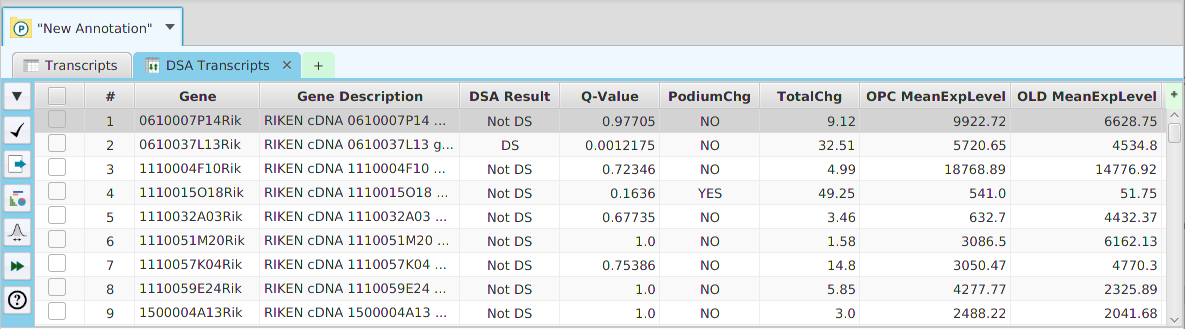
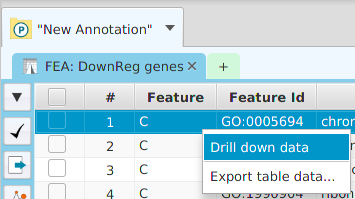
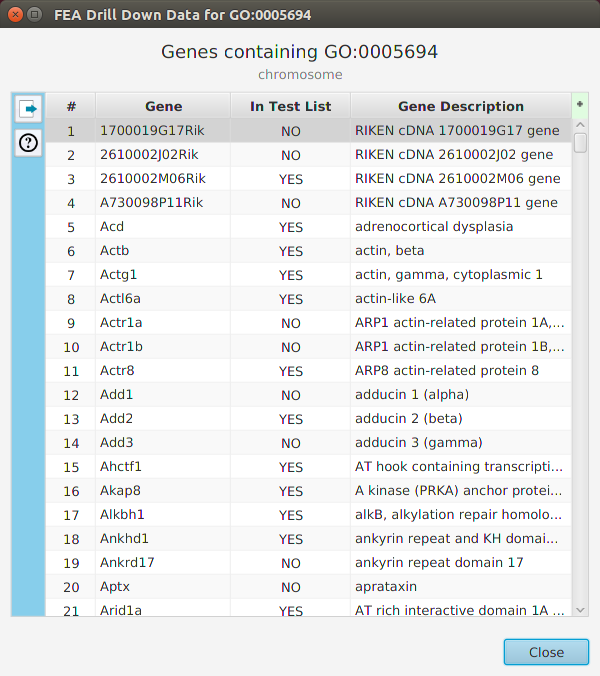
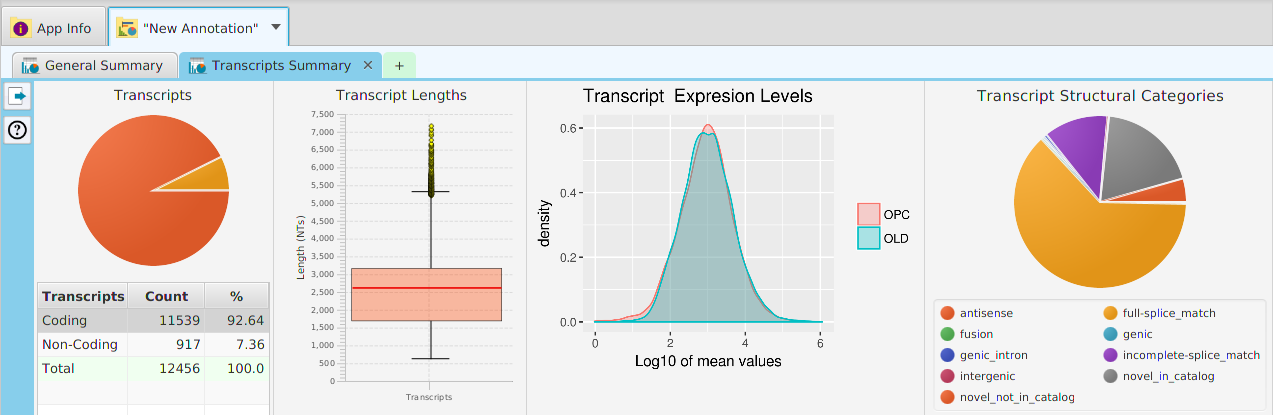
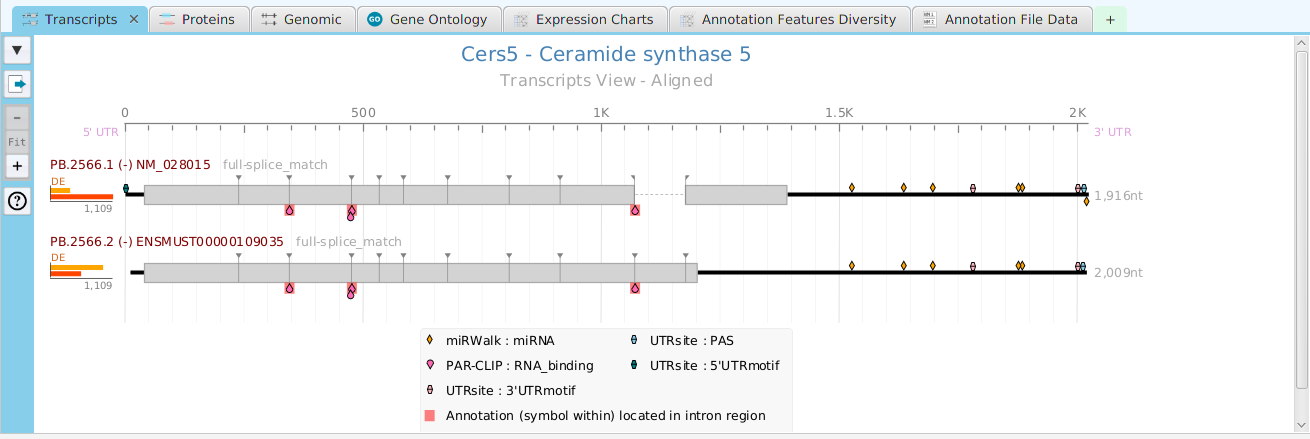
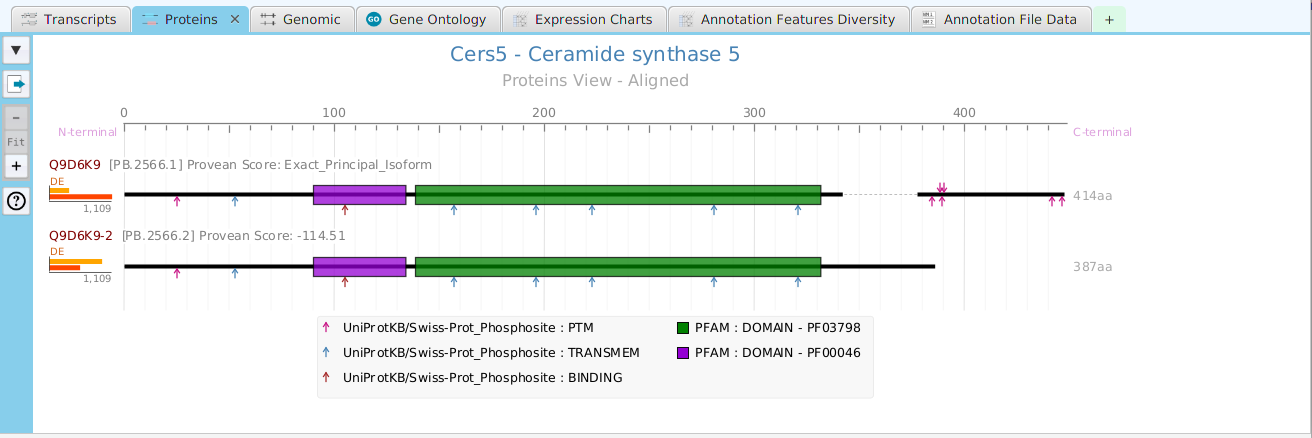
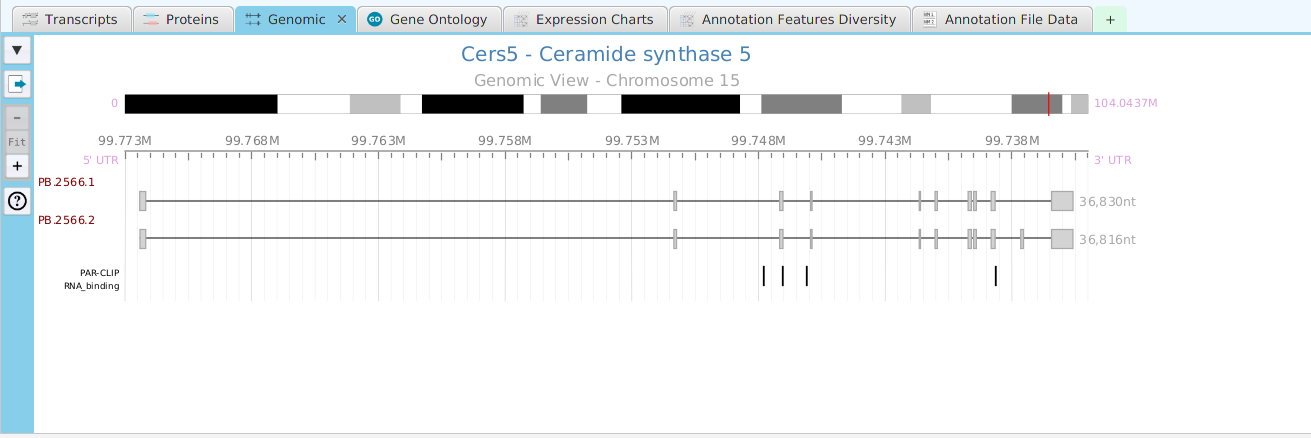
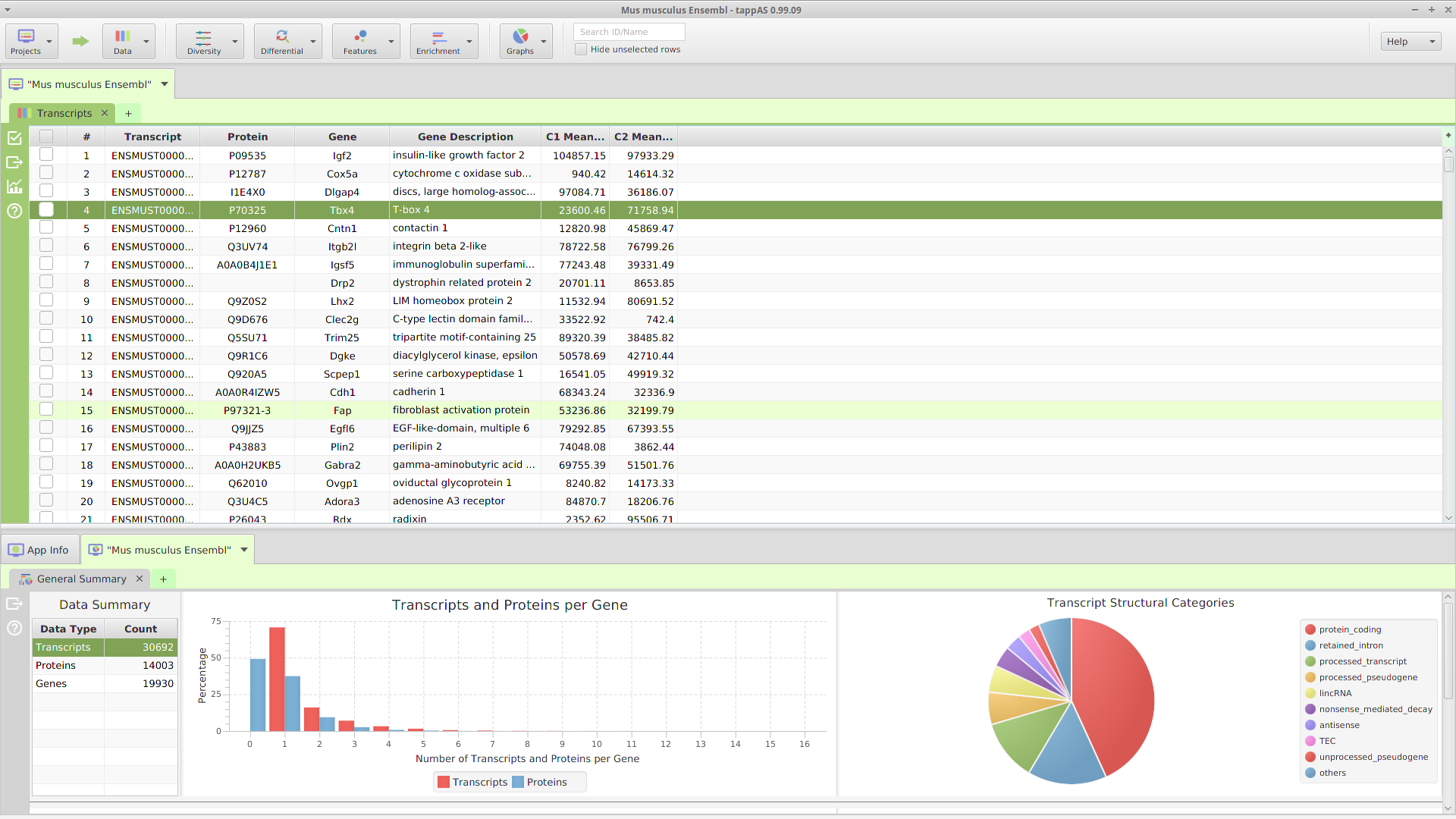
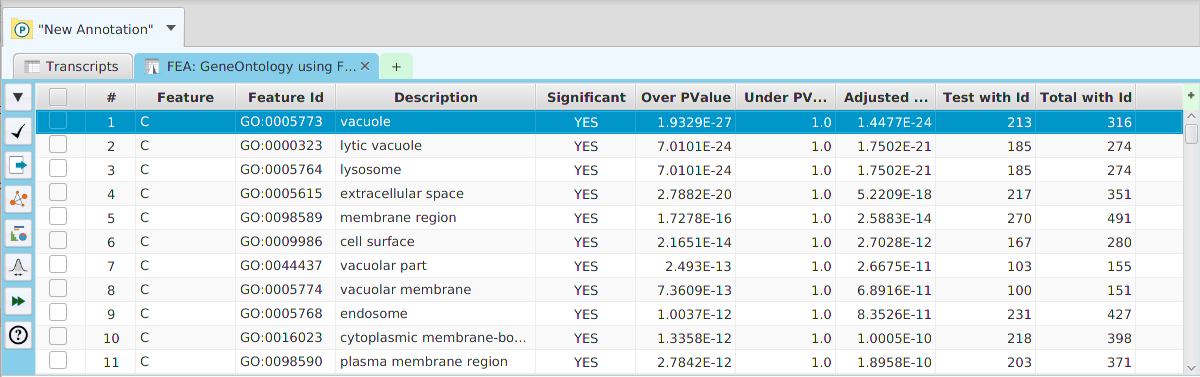
The FEA results are displayed in a table on the FEA Results subtab. The subtab is contained in the project data tab located in the top tab panel, see image below. Each table row displays the results, Significant – Yes/No, for a given annotation feature as well as over/under and adjusted P-Values. The number of test genes and total genes containing the feature are also shown.
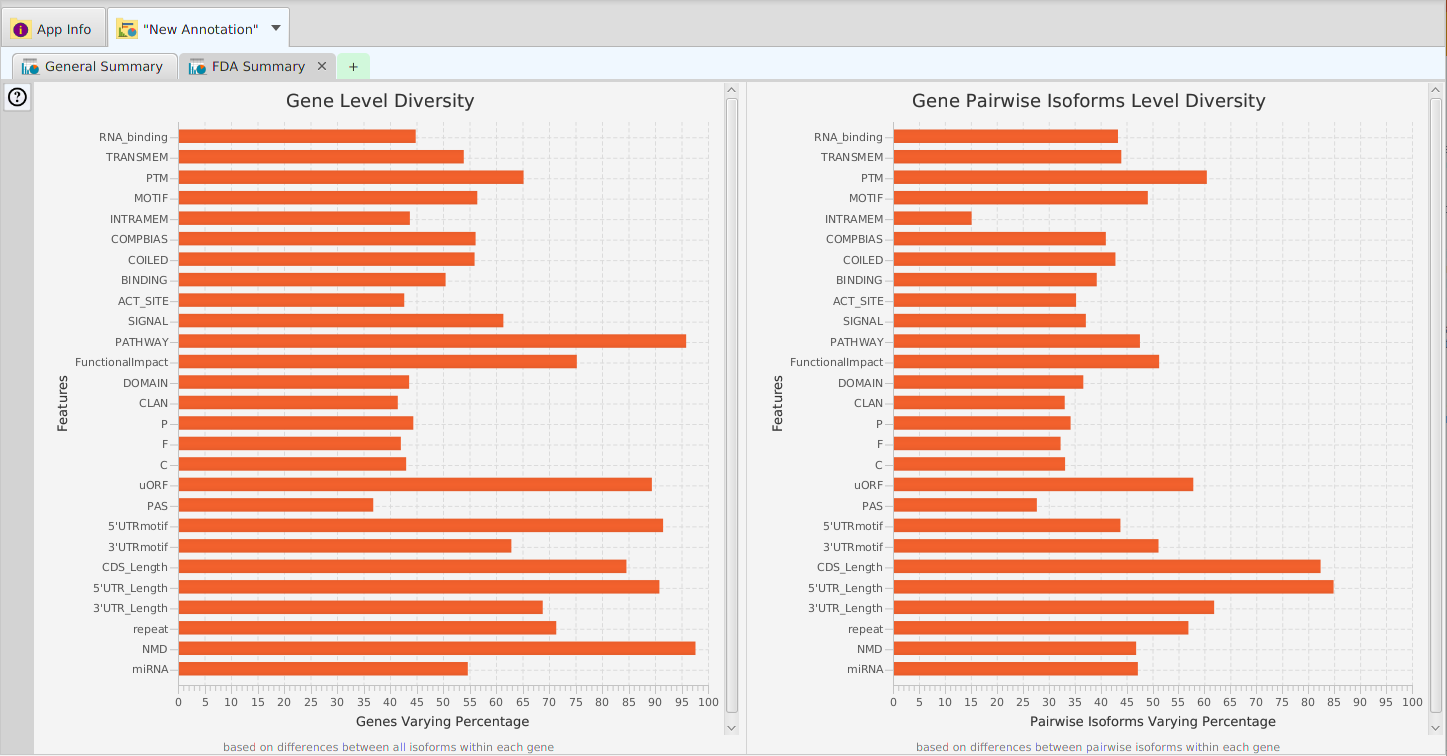
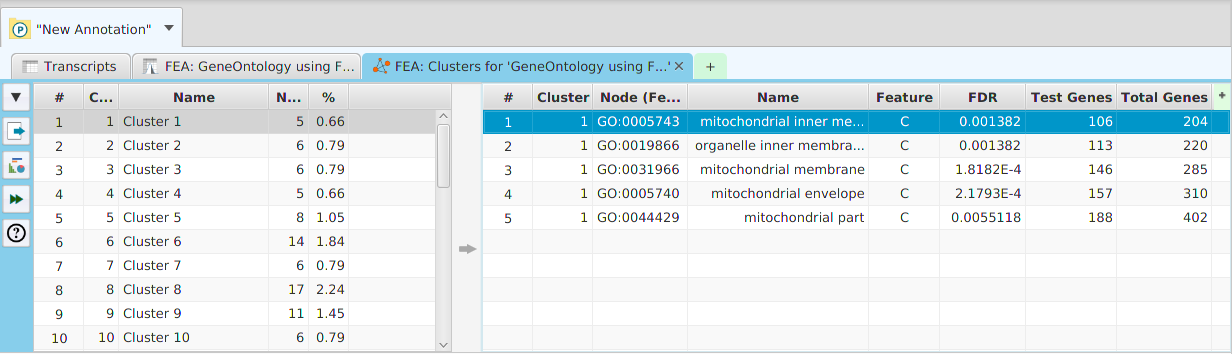
Enriched Features Cluster Analysis
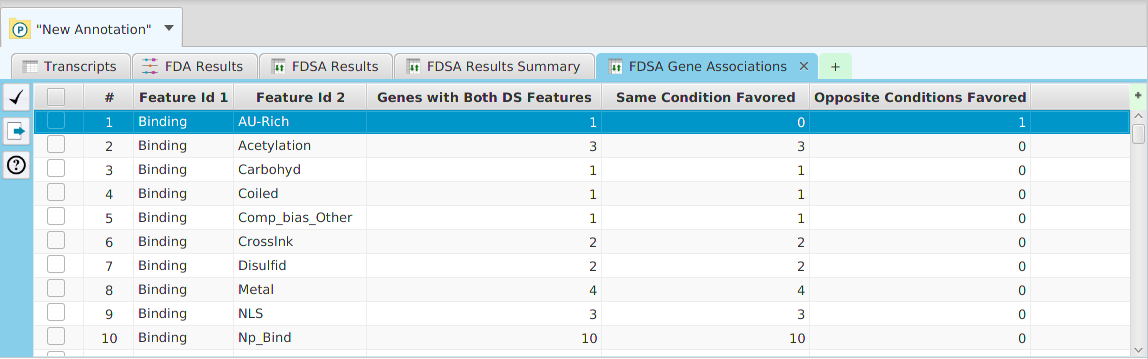
The option to run Cluster Analysis on the enriched features from the FEA results is provided. The cluster analysis results can be seen below. The table on the left displays the clusters while the table on the right displays the nodes for the selected cluster(s). You may select multiple clusters to see their combined nodes.
Gene Set Enrichment Analysis (GSEA)
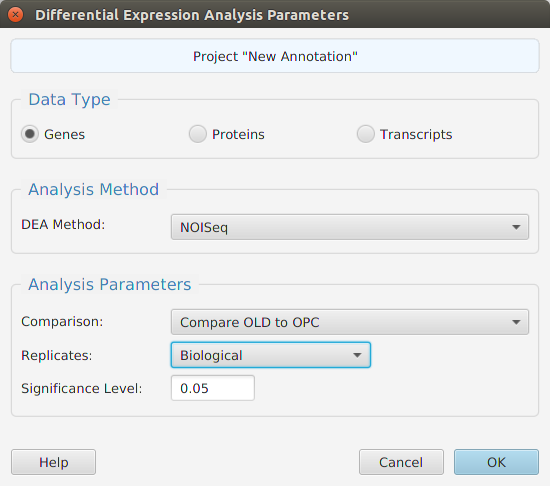
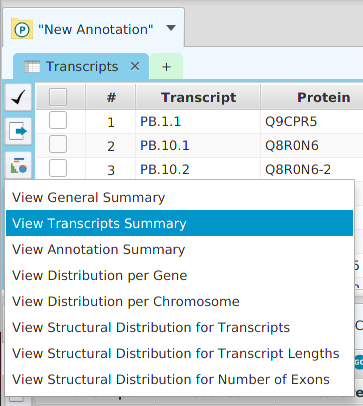
Gene set enrichment analysis… You may specify what expression data type to use – genes, proteins, or transcripts – for the analysis, and the corresponding ranked list to use. Available application ranked lists are provided but you may use any previously generated ranked list file. You also need to specify what annotation features to test for either by selecting from list or providing your own annotation sets in GMT file format.
Note: Will include content from the research paper, once it becomes available
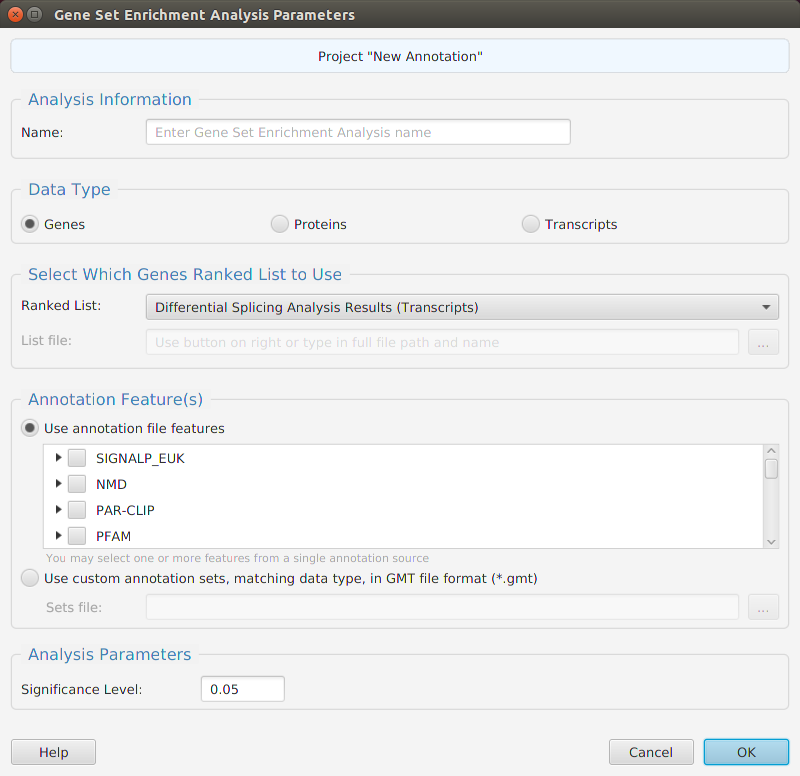
When using the application, all GSEA parameters are described in the Help page which can be accessed via the Help button located on the bottom left of the dialog window.
GOglm
GOglm provides “…”.You may view the documentation and installation instructions at:
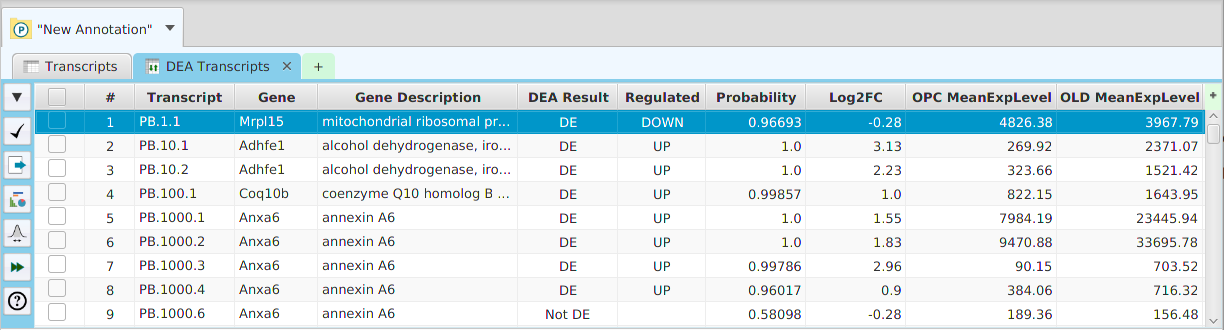
GSEA Results