Overview
Advances in RNA sequencing technology have made the reliable detection and quantification of gene expression at the isoform level possible. TAPPAS has been created specifically to provide analysis, visualization, filtering, and ad hoc query tools for working with RNA-seq data at the gene and isoform levels.Note: Most of this section will come from the research paper, once it becomes available
You can find the tappAS paper at Genome Biology:
De La Fuente, L., Arzalluz-Luque, Á., Tardáguila, M., Del Risco, H., Martí, C., Tarazona, S., Salguero, P., Scott, R., Lerma, A., Alastrue-Agudo, A., Bonilla, P., Newman, J. R. B., Kosugi, S., McIntyre, L. M., Moreno-Manzano, V., & Conesa, A. (2020). tappAS: a comprehensive computational framework for the analysis of the functional impact of differential splicing. Genome Biology, 21(1). https://doi.org/10.1186/s13059-020-02028-w
Application Requirements
tappAS is a Graphical User Interface (GUI) application written in Java. It uses SQLite for its database management and R, along with some statistical packages, for data analysis. Like most applications dealing with large datasets, the more computational power and memory available, the better the application will run. Be aware that insufficient resources, – CPU, memory, and disk space – will make the application sluggish or unusable. Listed below are the requirements for running TAPPAS:
Computer Hardware
- Minimum of 4 cores (multi-core CPU or single core CPUs)
- Minimum of 8 GBs of memory
- Minimum of 20 GBs of available, unused, disk space
Operating System
- Mac OS X recent version, or
- Windows OS recent version, or
- Linux Ubuntu recent version, or
- Any other Linux/Unix desktop environments able to run Java GUI applications should work but HAVE NOT BEEN TESTED
Software
- R and Rscript version 3.2.2 or later
R packages
- NoiSeq recent version
- maSigPro recent version
- edgeR recent version
- DEXSeq recent version
- goseq recent version
- GOglm version 0.4.2. or later
- ggplot2 recent version (CRAN Repository)
- VennDiagram recent version (CRAN Repository)
- MASS recent version (CRAN)
- plyr recent version (CRAN)
- ggrepel recent version (CRAN)
You can find more information about R packages in our install manual.
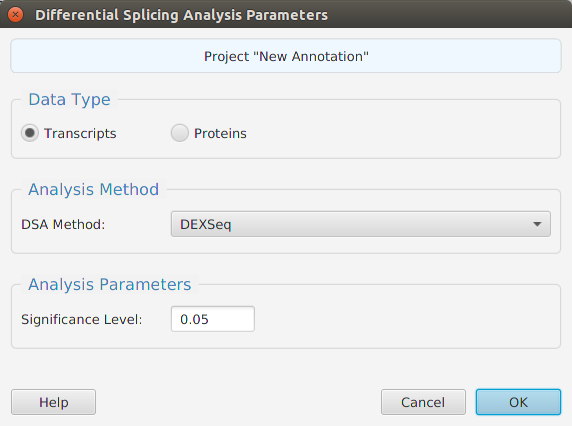
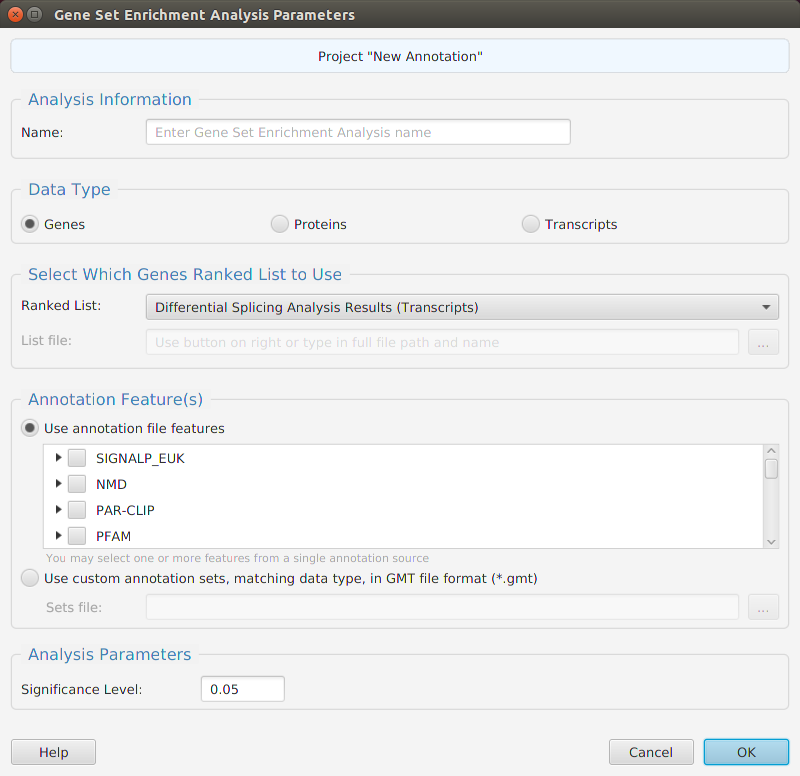
The tappAS application is project based: you create a project, input your data, and work with it. Each project has a corresponding file folder where all its data and analyses results are stored. All the necessary project management functions – create, open, rename, and delete – are provided in the application. To create a project, you must provide the following information:
- A unique project name
- The biological species associated with the RNA-seq data
- The file location for the annotation features file or select one of the application provided annotation files
- The experiment type
- The file location for your experiment design file
- The file location for your transcript level raw counts expression matrix file
- Optionally, but recommended, the low count and coefficient of variation filtering parameter
- Optionally, the inclusion or exclusion transcripts list file location for filtering
Input Data and Filtering
There are three input data files required to create a project: an experiment design file, a transcript level raw counts expression matrix, and a corresponding annotation file. The input data and optional filtering block diagram is shown below:
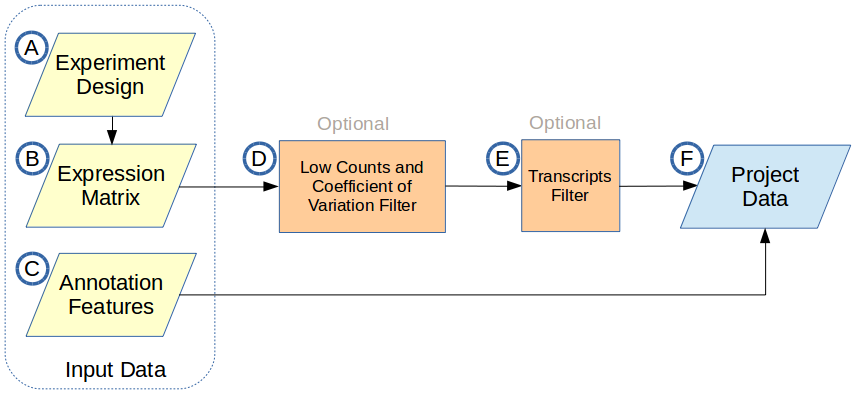
A. Experiment Design
B. Expression Matrix
C. Annotation Features
A data file containing annotation features for all expressed transcripts. Any transcript in the expression matrix that is not included in this file will be filtered out. You may use one of the annotation files provided by the application or use your own. The application currently provides the following annotation files:
- Homo sapiens – Ensembl and RefSeq
- Mus musculus – Ensembl and RefSeq
- Arabidopsis thaliana – Ensembl
- Zea mays – Ensembl
See Annotation Features File Format for details.
D. Low Counts and Coefficient of Variation Filter
E. Transcripts Filter
F. Project Data
Expression Matrix Data Normalization
As previously stated, the application keeps a copy of the original raw counts expression matrix and also creates a new matrix using normalized counts. The Trim Mean of M (TMM) normalization procedure by Robinson and Oshlack, provided in the R package NOISeq, is used to normalize the data.
You may view the NOISeq documentation and installation instructions at:
Experiment Design File Format
The experiment design file defines the relationship between the expression matrix data and the various experimental groups, time slots, and replicates. There are three experiment types supported by the application:
- Case-Control
- Time-Course Single Series
- Time-Course Multiple Series
The design file will change depending on the experiment type. However, regardless of experiment type, it is possible to use the same expression matrix and just modify the design file. By doing so, you have the option to run case-control analysis, and time-course single series analysis using the data from a time-course multiple series experiment. You may also, leave out replicates, time slots, etc. without having to make any changes to the expression matrix.
Regardless of what data you use from the expression matrix, the first experimental group is treated as the control group where relevant. The following format rules apply to all design files:
- The data must be in Tab Separated Values (TSV) format and must contain a single line header
- Comment lines are not allowed
- The first experimental group is considered the control group where relevant
- All samples for an experimental group must be grouped together
- All samples for a given time slot, within an experimental group, must be grouped together
- All time slots for a given group must be specified in chronological order
- Time values must be specified using numbers only – no time units
- Sample column names must be unique
- Sample column names are case-sensitive and must match the expression matrix
Case-Control Design File
The case-control design file must contain two experimental groups. Each group must contain at least two replicates.
Sample design file:
| sample | group |
|---|---|
| CASE1 | CASE |
| CASE2 | CASE |
| CONTROL1 | CONTROL |
| CONTROL2 | CONTROL |
Single Series Time-Course Design File
The single series time-course design file must contain a single experimental group. The group must contain at least two time slots with a minimum of two replicates per time slot.
Sample design file:
| sample | time | group |
|---|---|---|
| CASE1 | 0 | CASE |
| CASE2 | 0 | CASE |
| CASE3 | 3 | CASE |
| CASE4 | 3 | CASE |
Multiple Series Time-Course Design File
The multiple series time-course design file must contain at least two experimental groups. Each group must contain at least two time slots with a minimum of two replicates per time slot.
Sample design file:
| sample | time | group |
|---|---|---|
| CASE1 | 0 | CASE |
| CASE2 | 0 | CASE |
| CASE3 | 3 | CASE |
| CASE4 | 3 | CASE |
| CONTROL1 | 0 | CONTROL |
| CONTROL2 | 0 | CONTROL |
| CONTROL3 | 3 | CONTROL |
| CONTROL4 | 3 | CONTROL |
Expression Matrix File Format
The expression matrix file must contain raw expression counts for one or more experimental groups. Each group may have one or more time slots with each time slot having at least two replicates. The following format rules apply:
- The data must be in Tab Separated Values (TSV) format and must contain a single line header
- A unique transcript id identifies each row and must match one of the transcripts provided in the annotation file or it will be discarded
- Sample column names must be unique
- Sample column names are case-sensitive and must match the experiment design file
- The columns do not need to be in any specific order – the experiment design file will provide grouping information
Expression matrix file partial contents sample:
| NPC1 | NPC2 | OLD1 | OLD2 | |
|---|---|---|---|---|
| Transcript.1 | 7275 | 3602 | 3707 | 3485 |
| Transcript.2 | 358.64 | 206.58 | 2056.72 | 2094.65 |
| Transcript.2 | 332.44 | 329.38 | 1529.46 | 1318.57 |
| Transcript.4 | 46.92 | 13.03 | 20.82 | 33.78 |
Annotation Features File Format
The annotation file must follow the basic Generic Feature Format 3 (GFF3). However, it has been slightly modified to suit the application: the “score” and “phase” columns are not used and some of the attributes may not fully abide by the formal specifications. The file consists of a set of annotation features for each transcript. Each set of features is divided into sections as follows:
Transcript Level Feature Annotations – basic transcript information, UTR motifs, microRNAs, etc.
Genomic Level Feature Annotations – exons, splice junctions, etc.
Protein Level Feature Annotations – gene ontology features, domains, phosphorylation sites, etc.
Transcript 2
…
Transcript 3
…
Some of the annotation features must be named as expected by the application, see sample annotation file below:
| Source | Feature | Description |
|---|---|---|
| tappAS | transcript | Start of transcript features |
| tappAS | gene | Gene information |
| tappAS | CDS | CDS information |
| tappAS | genomic | Start of genomic features |
| tappAS | exon | Exon |
| tappAS | splice_junction | Splice junction |
| tappAS | protein | Start of protein features |
In addition, the following attributes must be named as required by the application, see sample annotation file below:
| Attribute | Description |
|---|---|
| ID | Feature ID |
| Name | Feature name |
| Desc | Feature description |
| Chr | Feature chromosome |
Annotation file partial contents sample (header should not be included):
| SeqName | Source | Feature | Start | End | Score | Strand | Phase | Attributes |
|---|---|---|---|---|---|---|---|---|
| PB.3189.4 | tappAS | transcript | 1 | 1399 | . | + | . | ID=XM_006524897.1; primary_class=full_splice_match; PosType=T |
| PB.3189.4 | tappAS | gene | 1 | 1399 | . | + | . | ID=Qpct; Name=Qpct; Desc=glutaminyl-peptide cyclotransferase (glutaminyl cyclase); PosType=T |
| PB.3189.4 | tappAS | CDS | 10 | 951 | . | + | . | ID=XP_006524960.1; PosType=T |
| PB.3189.4 | UTRsite | 3’UTRmotif | 1288 | 1295 | . | + | . | ID=U0023; Name=K-BOX; Desc=K-Box; PosType=T |
| PB.3189.4 | UTRsite | PAS | 1380 | 1399 | . | + | . | ID=U0043; Name=PAS; Desc=Polyadenylation Signal; PosType=T |
| PB.3189.4 | mirWalk | miRNA | 986 | 993 | . | + | . | ID=mmu-miR-495-5p; Name=mmu-miR-495-5p; Desc=UTR3; PosType=T |
| PB.3189.4 | tappAS | genomic | 1 | 1 | . | + | . | Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | exon | 79052257 | 79052388 | . | + | . | Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | exon | 79070673 | 79070951 | . | + | . | Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | exon | 79077482 | 79077658 | . | + | . | Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | exon | 79079467 | 79079566 | . | + | . | Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | exon | 79081747 | 79081863 | . | + | . | Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | exon | 79089623 | 79090216 | . | + | . | Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | splice_junction | 79052388 | 79070673 | . | + | . | ID=known_canonical; Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | splice_junction | 79070951 | 79077482 | . | + | . | ID=known_canonical; Chr=chr17; PosType=G |
| PB.3189.4 | tappAS | splice_junction | 79077658 | 79079467 | . | + | . | ID=known_canonical; Chr=chr1; PosType=G |
| … | … | … | … | … | … | … | … | … |
| PB.3189.4 | tappAS | protein | 1 | 313 | . | + | . | ID=NP_001303658.1; PosType=P |
Generating an annotation file is not a trivial task and it’s not recommended unless you have a good programming background and knowledge of annotation features. If possible, use one of the annotation files provided by the application. If no annotation file is provided for the species you are interested in, you may contact us .
tappAS is a Java application and its Graphical User Interface (GUI) is based on JavaFX. Using JavaFX allows the application to work across multiple Operating Systems (OS) and provide the same look and feel of native applications. In addition, JavaFX allows the application to provide the rich set of features expected from a modern GUI application.
GUI Layout
The application layout consists of 3 main sections: a top tool bar and two tab panels, a data tab panel on top and a data visualization tab panel on the bottom, see image below.
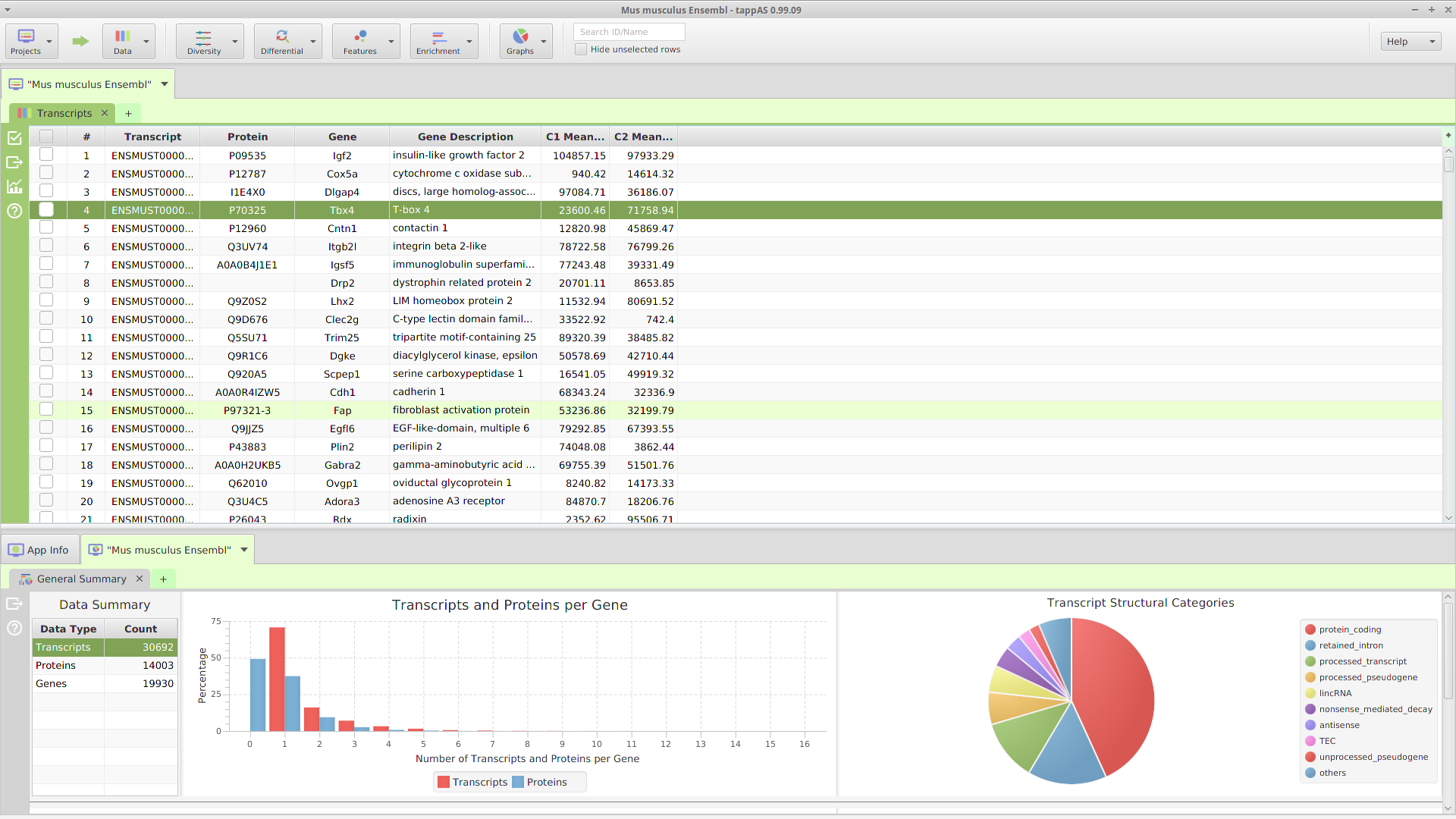
Application GUI Layout
A. Top Tool Bar
The top tool bar provides access to all the high level functionality in the application. Starting on the left, it contains multiple menu buttons:
- Projects – provides access to all the project management functions: create, open, close, list, and delete
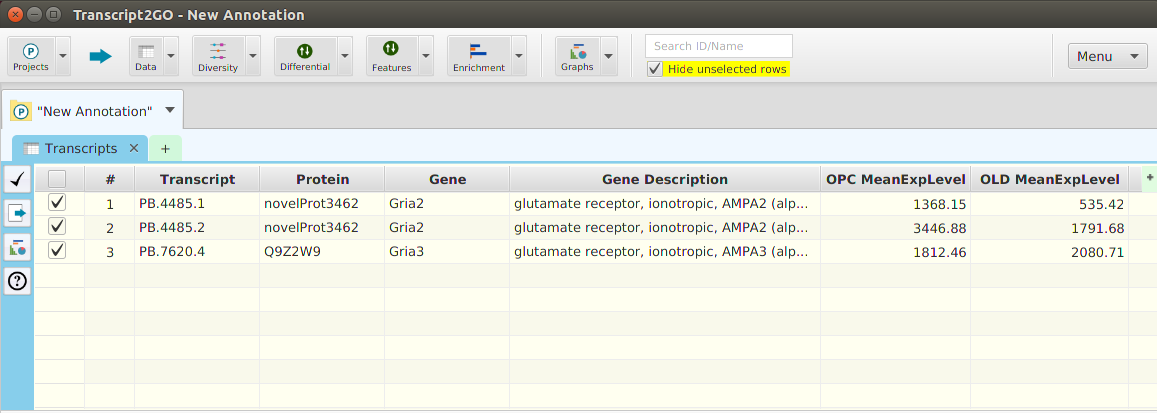
- Data – provides access to all the project data: transcripts, proteins, genes, and original expression matrix. In addition, it provides a menu selection to reinput the project data
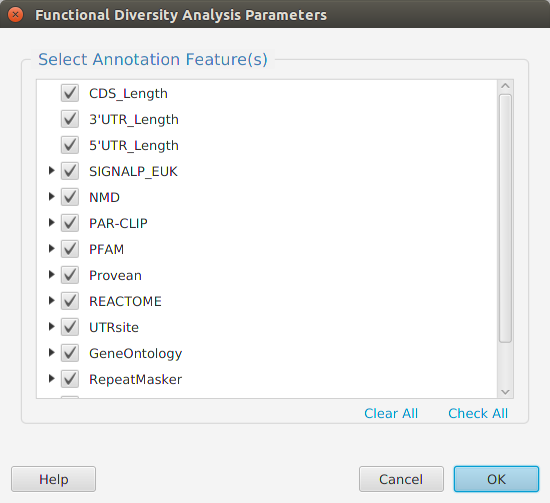
- Diversity – provides access to all the annotation features diversity management functions: run analysis, view and clear analysis results
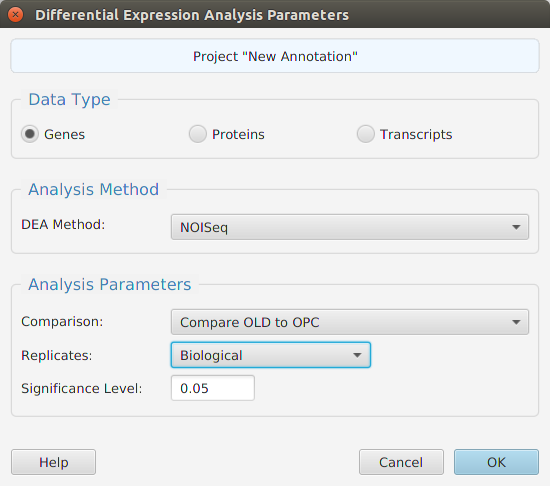
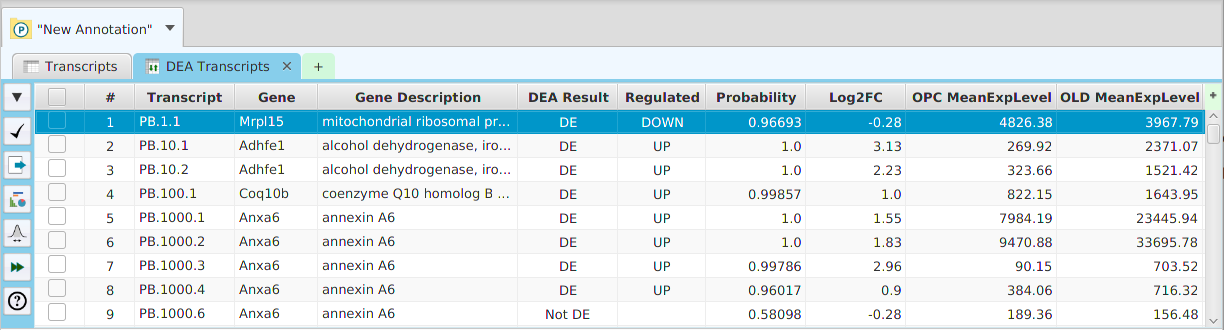
- Differential – contains all the differential expression and splicing analysis management functions: run analysis, view and clear analysis results
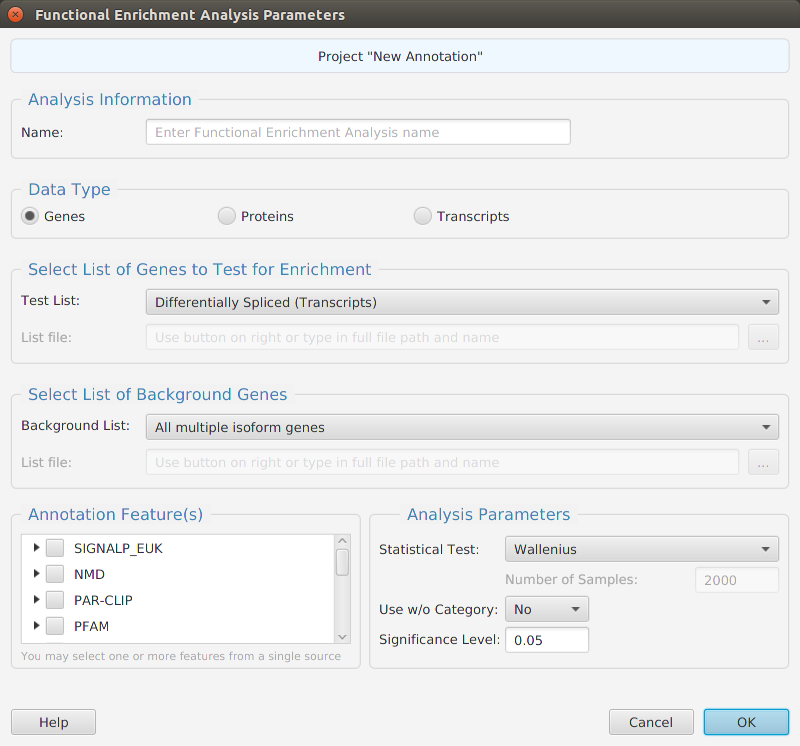
- Features – contains all the enrichment analysis, FEA and GSEA, management functions: run analysis, view and clear analysis results
Located after the menu buttons, are the data table search text field and the filter checkbox controls. These controls apply to the currently selected data table, in one of the subtabs below, and as their name implies, are used for searching and table row filtering purposes. Finally, all the way on the left, there is a menu button to access miscellaneous application functions.
B. Top (Data) Tab Panel
C. Bottom (Data Visualization) Tab Panel
D. Data Tab
E. Data Visualization Tab
F. Gene Data Visualization Tab
G. Annotation Source Tab
H. Application Tab
I. Subtab
J. Subtab Menu Bar
Tabs and subtabs will be discussed in details in the Tabs section.
Context-Sensitive Menus
In addition to all the visible menu buttons in the application, there are context-sensitive menus all over the application that are not visible. Context-sensitive menus are popup menus that are only shown as a result of a right-click with the mouse on a user interface display element. The menu item selections shown, and/or the actual data displayed when a selection is made, will vary based on what display element, or even what part of it, was right-clicked. For example, gene data visualization is accessed via context menus, what gene the data visualization is shown for depends on what row of the data table the right-click took place on, see image below. The same row specific context display applies to drill down data displays.
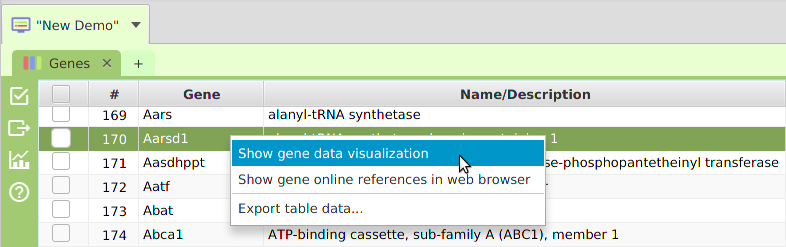
Gene Context-Sensitive Menu
Application functionality can sometimes be accessed more efficiently via context menus. For example, if you have multiple display elements on a data visualization subtab, you may right-click on the display element you are interested in and the export menu selection shown in the context menu will be exclusively for that element. The gene data visualization and drill down data, previously mentioned, are examples of functionality that is only accessible via context menus. Make sure to not miss out on application functionality accessible only in context menus: when in doubt, right-click and see what pops up.
Tab Panels, Tabs, and Subtabs
All application information display is organized into tab panels, tabs, and subtabs. The tabs, depending on their type, are displayed by default in either the top or bottom tab panels, see Application GUI Layout image. However, before we proceed, let’s review the terminology:
- Tab panel – refers to a display control that contains tabs
- Tab – refers to a display control, contained in a tab panel, that contains subtabs
- Subtab – refers to a display control, contained in a tab, where the actual information display takes place, i.e. tables, charts, etc.
And the display hierarchy is:
There are five different types of tabs in the application:
- Project Data Tab (one per project) – contains project data and analysis result subtabs and is displayed on the top tab panel by default
- Project Data Visualization Tab (one per project) – contains project data visualization subtabs and is displayed on the bottom tab panel by default
- Gene Data Visualization Tab (one per gene, project specific) – contains all gene data visualization subtabs, see Gene Data Visualization section for details, and is displayed on the bottom tab panel by default
- Annotation Source tab (one per project) – contains annotation features details and data visualization subtabs for selected annotation source and is displayed on the bottom tab panel by default
- Application Tab (one per application) – contains global application information subtabs such as the log, overview, technical information, etc. It is displayed on the bottom tab panel by default
The gene data visualization tab and the application tab display a relatively small number of subtabs. However, the project data and data visualization tabs can display a significant number of subtabs for all the data, analysis results, and corresponding data visualization. It will be up to you to explore the application and see all that’s available.
Subtabs Menu Bar
A significant amount of your interaction with the application will take place via the subtabs menu bar, see Application GUI Layout image. It contains a set of menu buttons to provide the functionality required based on the subtab contents. You can take advantage of the mouseover functionality available for all buttons, to find out what it does, or just click on it to find out. The application will always confirm your request before doing anything destructive so have no fear. Once you can associate the button images with their functionality, the application becomes easier to use. The subtab menu bar buttons along with their respective functionality are:
|
|
– miscellaneous options menu will change based on subtab content |
|
|
– table row selection management menu |
|
|
– export data or images menu |
|
|
– data visualization menu |
|
|
– clustering analysis menu |
|
|
– rerun analysis |
|
|
– change analysis significance level |
|
|
– show subtab help |
|
|
– zoom control buttons |
Tables
All application tables use a standard GUI so you should be familiar with basic functionality like scrolling, resizing columns, etc. There are some features you may not be familiar with:
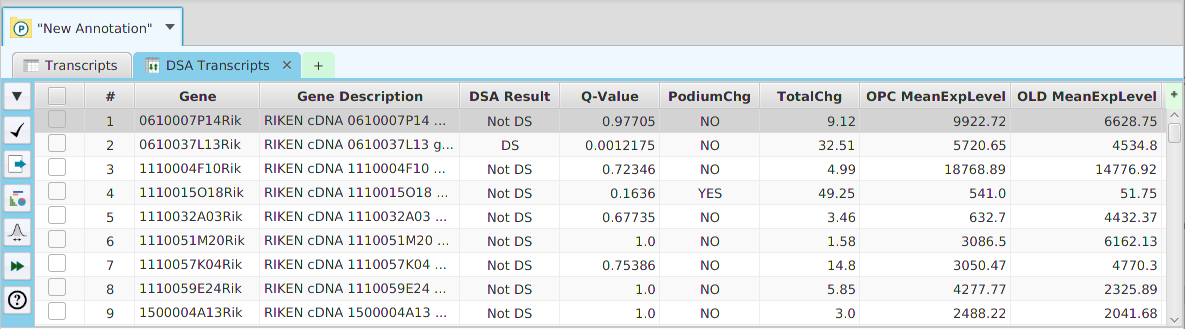
- Column sorting – if you click on a column header (where the column name is displayed) you can sort the table rows based on the contents of that column. If you click on the same column header multiple times you go through a cycle: ascending sort, descending sort, and clear sort. You may also sort by multiple columns. To do that, you click on the first column you want to sort by and then you hold the shift key and click on the next column you want to sort by. An example would be to sort by the DSA Results column in the DSA results table and then shift-click on the Q-Value column to see them in order.
- Show/hide columns – if you look at the top right corner of the table, you will see a small plus sign on a green background. If you click on it, a drop down menu will appear, see table image below. Each column will be displayed as a menu selection and the columns currently shown will have a check mark by them while the ones that are not shown will not. You may toggle the show/hide status by clicking on the column menu selection. If applicable, depends on the table, you may also add special annotation feature columns, on a need to basis, using the “Add annotation feature column…” menu selection at the bottom. You should only add annotation feature columns if you intend to use them for filtering. If you add the feature name/description column, be aware that some annotation features have long descriptions, such as GO terms, and can use up a considerable amount of memory.
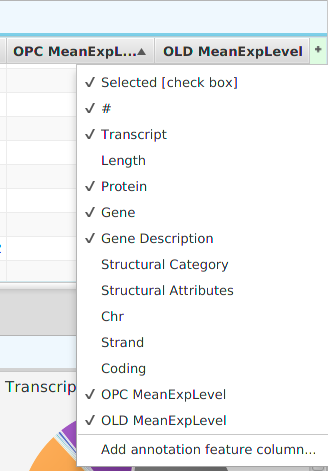
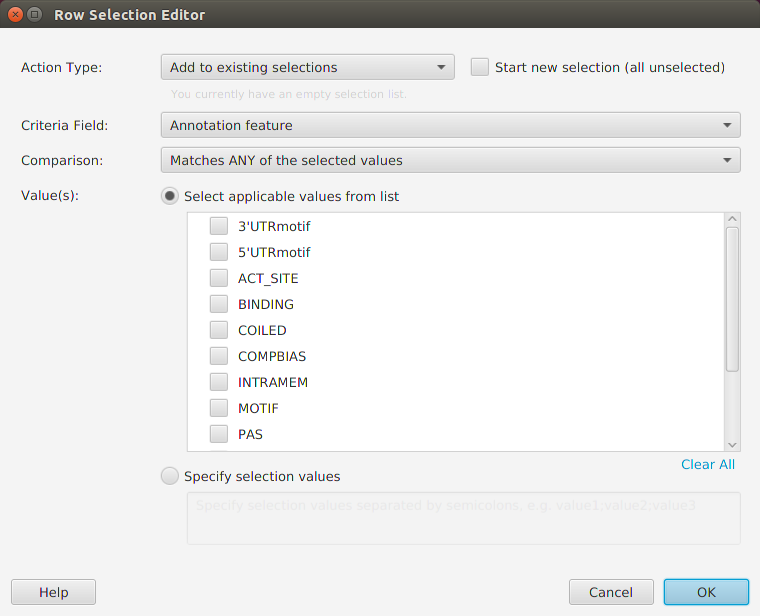
You may export the table data to file via the export menu button on the subtab bar or via the table’s context menu. Table search and row selection functionality is covered in the Ad Hoc Query section.
Visual Display Controls
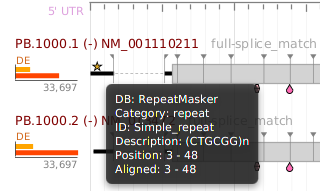
Most visual display controls – charts, graphs, etc. – in the application provide some interactive functionality:
- Mouseover – if you hover the mouse pointer over some areas, additional information will be displayed in the form of a tooltip. For example, if you hover the pointer over a pie chart section, it will normally display the section name and count/percentage information
- If you right-click on the control, a context menu will popup and provide an export image menu selection
There are some special visual display controls that provide additional functionality for customizing or interacting with the display:
Annotation features visualization controls
In the Gene Data Visualization tab, there are 3 special annotation features visualization controls in the transcript, protein, and genomic subtabs. In addition to providing the basic functionality previously mentioned, they also support:
The Options button in the Subtab Menu Bar section provides multiple options to customize the display and filter the data shown:
- Show gene isoforms aligned or unaligned
- Show/hide splice junctions (only if aligned)
- Show/hide PROVEAN score (proteins only)
- Show/hide ruler
- Show/hide display of structural attributes
- Sort isoforms by various methods
- Show only varying annotation features (varying among isoforms)
- Filter annotation features displayed
Note: some options are not applicable to all 3 subtabs and will not be available in all menus
Horizontal Zoom
If you double-click on the display, it will zoom in. If you hold the shift key down and double-click on the display, it will zoom out. Given the nature of the display contents, zooming only affects the horizontal axis. The same functionality is provided in the subtab bar using the zoom buttons, see Subtab Menu Bar section.
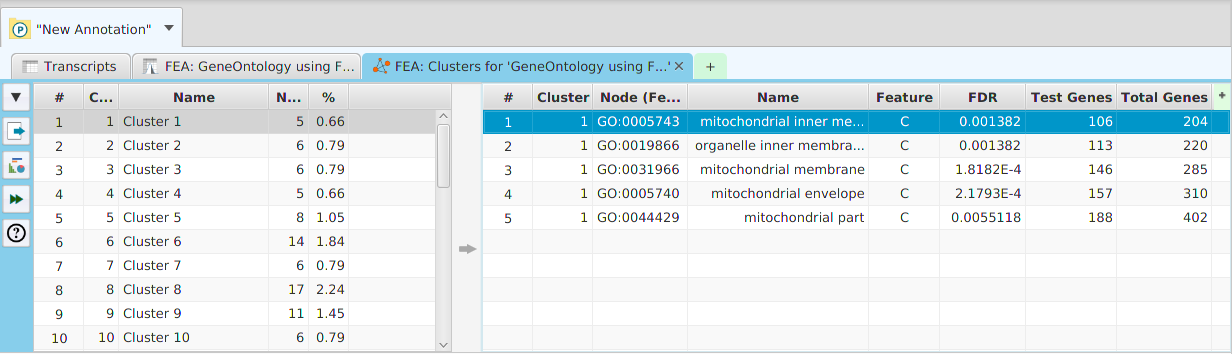
Network clusters and GO terms graph controls
The network clusters graph, and the GO term directed acyclic graph, support zooming in/out by clicking and also support panning:
If you double-click on the display, it will zoom in. If you hold the shift key down and double-click on the display, it will zoom out. You may also use the mouse scroll wheel to zoom in and out.Pan
Panning refers to ‘dragging’ the display area around with the mouse. It is typically done by pressing the left mouse button button down, on an empty area of the display, and keeping it down while moving the mouse around to ‘drag’ the display area.
Here are some tips to help you get the most out of the application:
- Use context-sensitive menus – don’t be afraid to right-click on any display control and see what pops up
- Take advantage of mouseover functionality – hover over buttons and visual display areas to see tooltips with additional information
- All subtabs have a menu bar on the left, explore the functionality provided in the menu buttons, see Subtab Menu Bar section
- Use the search text box on the top tool bar to search tables – search typically only includes Id, name, and description fields

- Remember table columns can be clicked for sorting or right-clicked for row selection
- Don’t forget tables can have additional columns that are not shown by default; use the table menu, green + button on the top right corner of the table, to show/hide columns
- The ability to filter tables, using row selection, and export filtered lists to use as input for data analysis is a powerful tool – take advantage of it
- The background color of tables and some display controls will change from white to yellow to indicate the data has been filtered
- Subtabs and dialog windows provide a help button for accessing detailed help information, use it as needed


- If the ‘See App Log’ notification is displayed on the top right section of the application tool bar, see image below, an error has occurred.
Click on the notification symbol, or manually select the application log subtab, to see the error message. Read the error message: you may be able to figure out what if any corrective action should be taken.
- The more tabs and subtabs you keep opened, the more memory resources the application uses
- Running the application on computers low on disk space, with insufficient memory, or totally overloaded with opened applications, will eventually lead to errors
Data visualization is a powerful tool for recognizing patterns, detecting correlations, and better understanding the data. TAPPAS provides a diverse set of visual elements for this purpose:
- Summary graphs, charts, and plots
- Distribution charts
- Annotation features visualization graphs for gene, proteins, and transcripts
- Expression level data density and PCA plot
- Cluster network graphs
- GO terms directed acyclic graphs
- Venn diagrams
- Other miscellaneous visualization displays
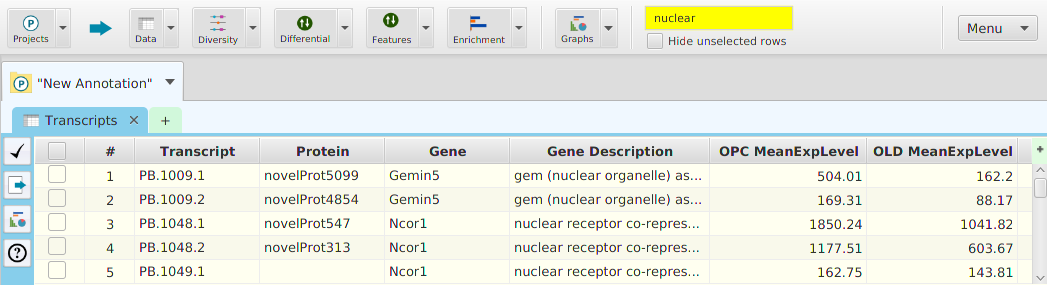
Accessing Data Visualizations
Data visualization display subtabs are provided for most data tables in the application. The easiest way to access data visualization for a specific table is to click on the data visualization button provided in the data subtabs and then choose from one of the menu item selections, see image below. Alternatively, you may use the Graphs menu button on the application’s top tool bar and select accordingly.
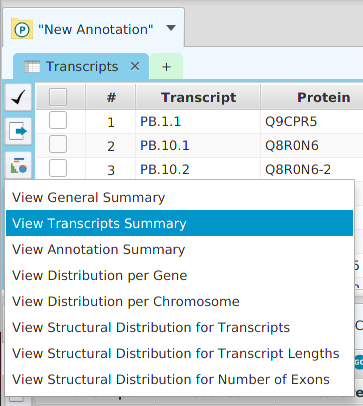
Once you make a selection, the data visualization subtab will be shown in the project’s data visualization tab, see image below.
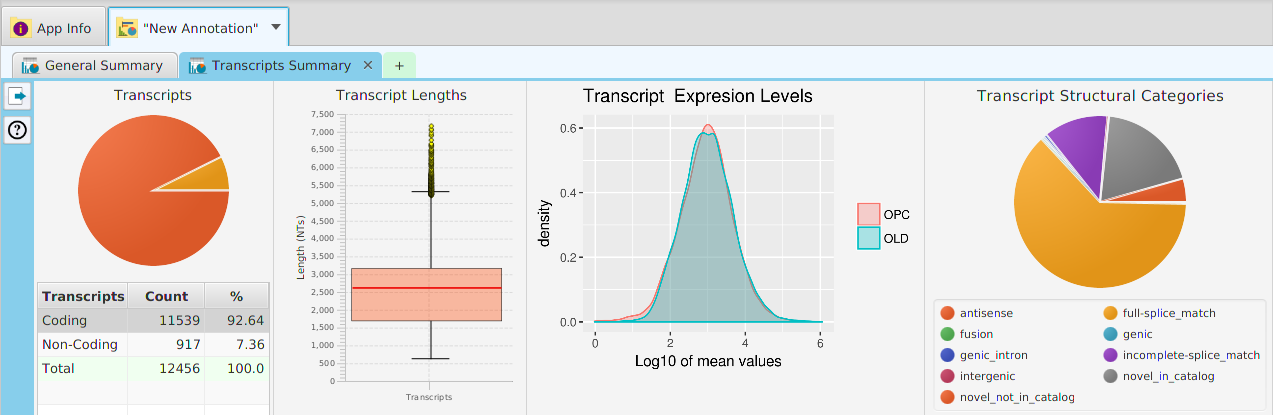
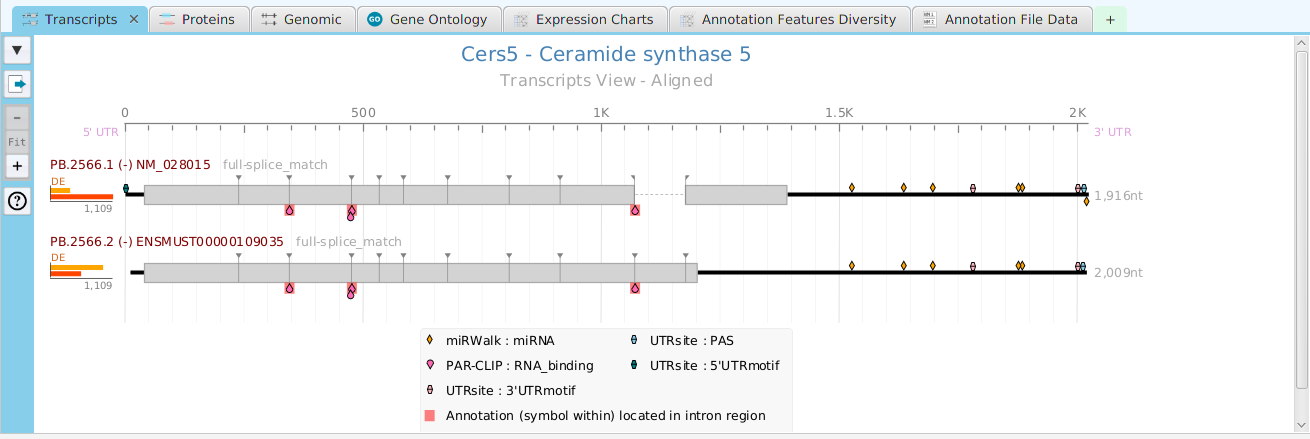
Gene Data Visualization
tappAS provides a self contained display tab for gene data visualization. It includes a comprehensive set of data visualization subtabs for gene annotation features down to the individual isoforms. The following subtabs are included:
- Transcripts – display of transcript annotation features
- Proteins – display of protein annotation features
- Genomics – full genomic view showing exons, introns, and genomic annotation features
- Gene Ontology – display of gene ontology graph for GO annotation features
- Expression Charts – display of expression level charts for gene, proteins, and transcripts
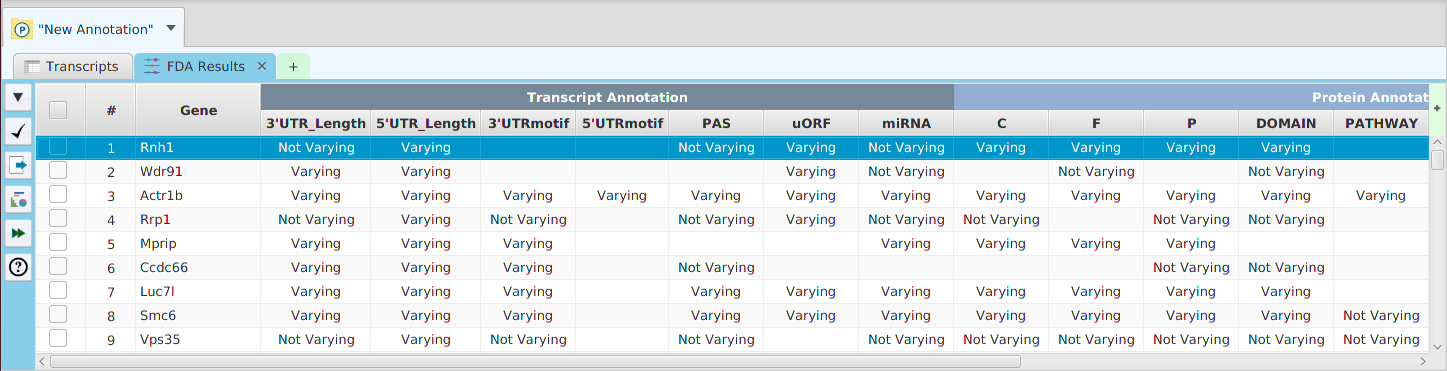
- Annotation Features Diversity – cross table display of annotation features and transcripts/proteins
- Annotation File Data – display of all annotation features for this gene contained in the annotation file
To access the visualization data for a specific gene, right click on the table row containing the gene of interest, for example the gene data table or the DIU results table, and click on the ‘Show gene data visualization’ menu item selection in the context menu. See Context-Sensitive Menus section. You may use the slide control buttons below to see all gene data visualization subtabs snapshots.
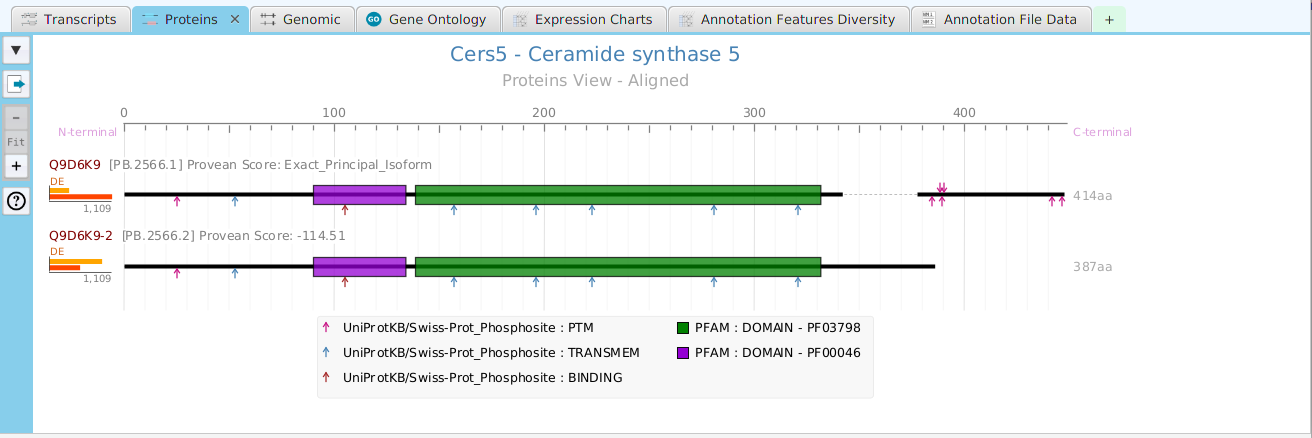
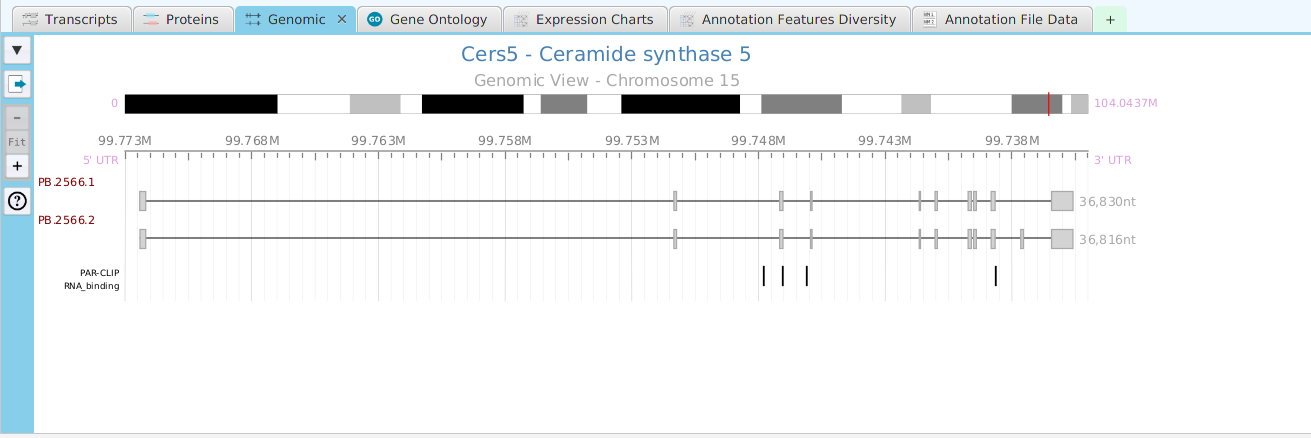
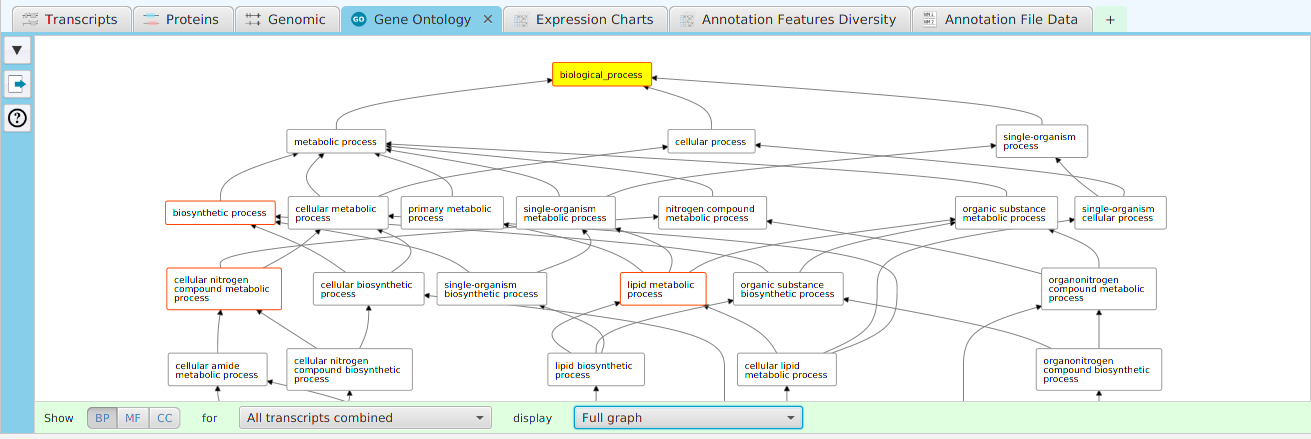
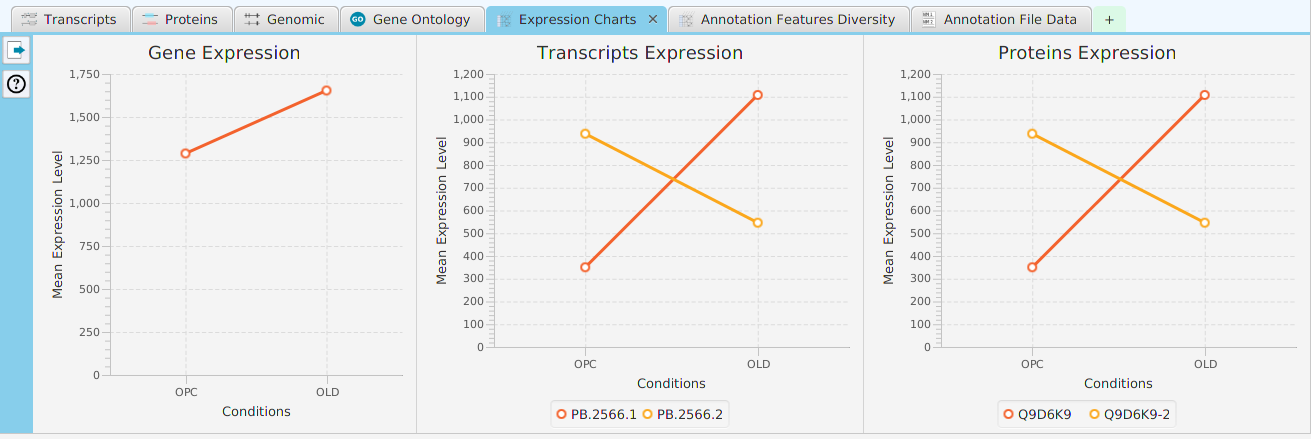
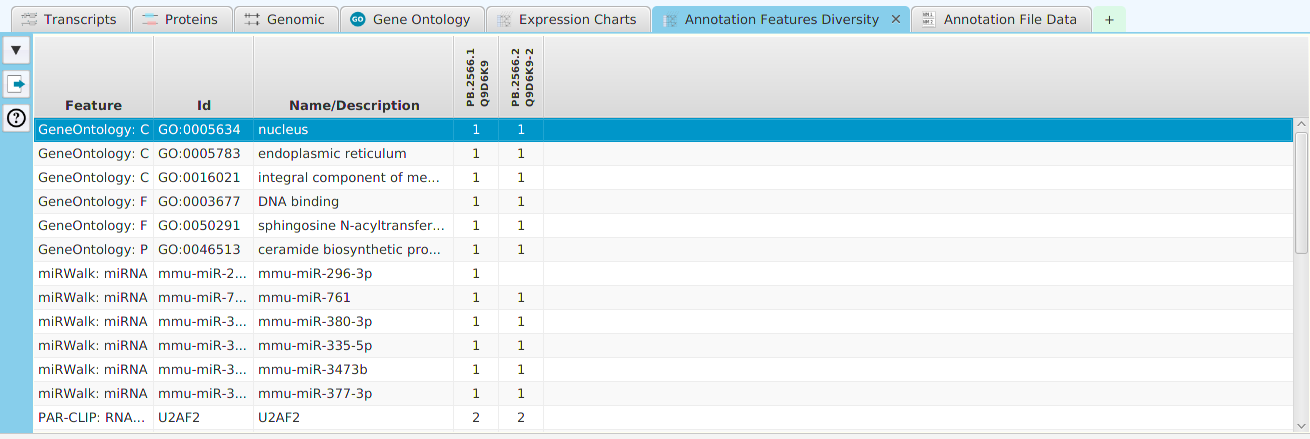
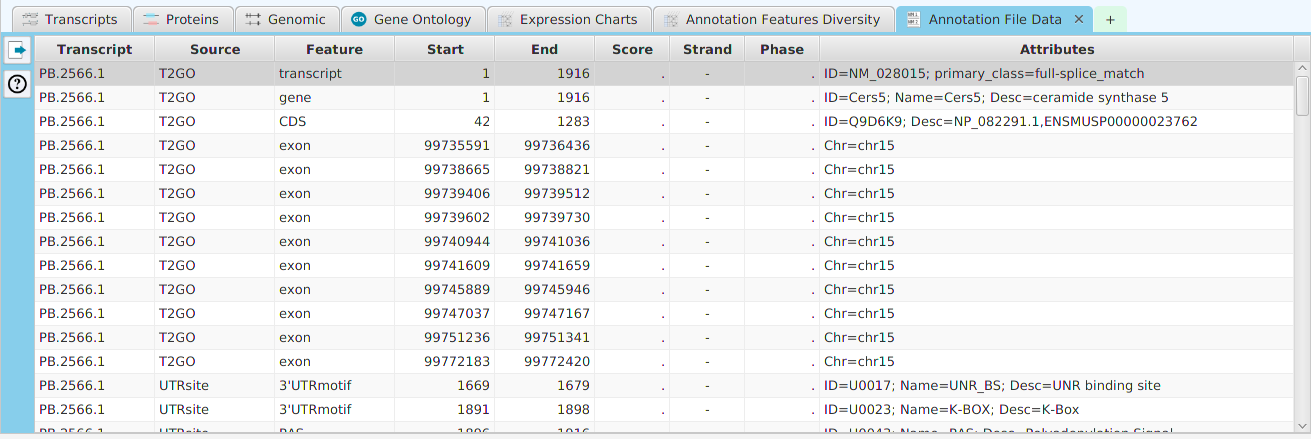
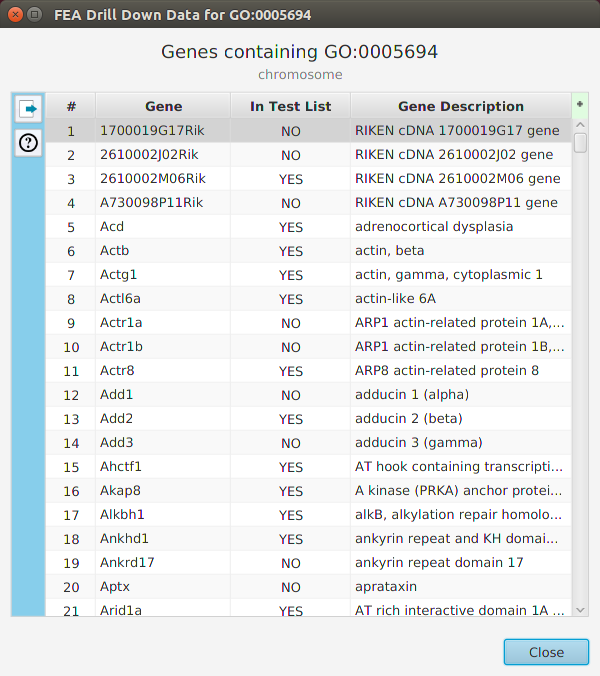
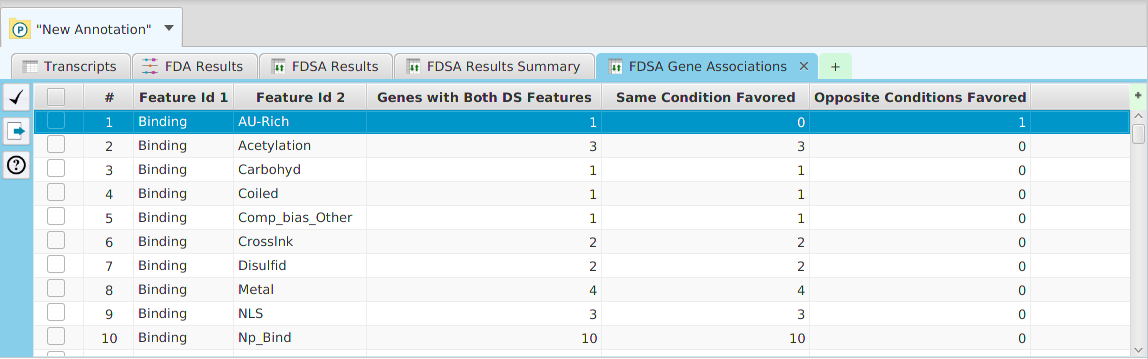
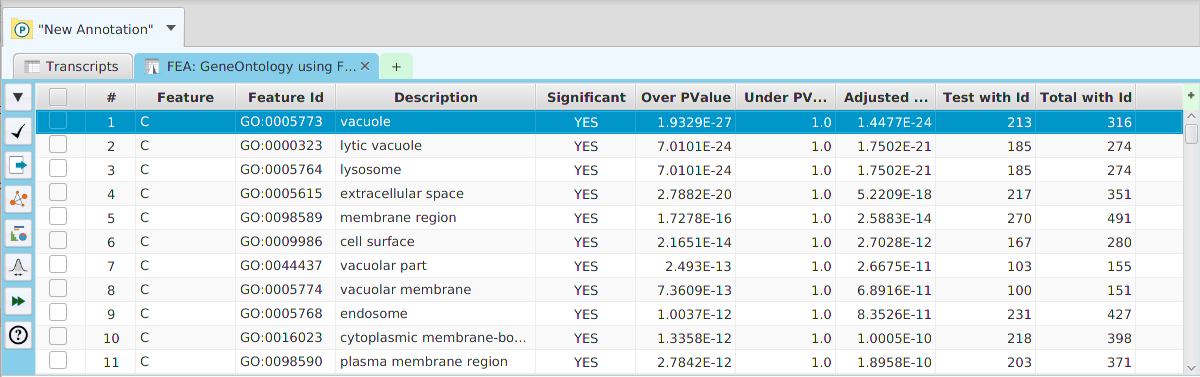
The ability to see the underlying data details can be extremely helpful and is provided, where relevant, via context-sensitive menus. As previously discussed in the Context-Sensitive Menussection, the data table row that you right-click on will determine the contents of the drill down data. For example, in the FEA results for Gene Ontology features window, shown below, the context menu provides a selection to drill down data.
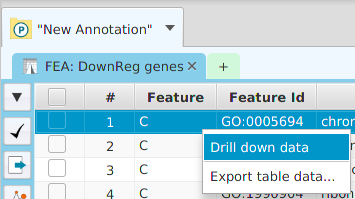
Once selected, the drill down data window will be displayed, see image below. Note the drill down data is for “GO:0005694” which is the selected table row. You may export the drill down table data and, for this specific example, view gene data visualization for specific genes via context menu.